Cleaning up 5 years of genomics work in 36 hours
Automating an unwieldy genomics processing & analysis pipeline with Snakemake and friends.

In September 2021, I spent the last 36 hours on a rollercoaster made of snakemake, {Rmarkdown}, {renv}, and conda/Docker. The ending had me think I might become the Joker. But the journey was fun.
What we were doing
@Laura_Kellman spent the last couple of years working on a cutting-edge cancer genomics project, accumulating many gigabytes of files and writing tens of RMarkdown notebooks. I volunteered to do a code review and functional replication on another machine.
Step 0: Gather everything
Where do you start when you want to do this? Gather everything.
- Make a folder.
- Gather data. If you have any idea where the data came from, great! If you don’t, stick them in the pile and we’ll figure it out later! Put them all in the
data/subfolder. - Gather code.
- Gather software requirements. What do I need to do to re-create your laptop’s computational environment?
- Bake a cheesecake. It will help you in your moments of anguish.
Step 1: Version control everything, including large data files
To be able to track any changes to the scripts, as well as any alterations to the data generated, we initialized a git repository in RStudio and threw everything in it. And I mean everything, including - perhaps against best practice - data files of up to 1 GB.
(Yes, it would have been better to install git-lfs or git-annex. You certainly cannot do this with really big data, but we decided everything under 1 GB was fair game, despite RStudio’s heartfelt protests. The idea is that this repository is perhaps not the version history we’ll share, but rather a safety net beneath our efforts.)
Note that if you do this, you cannot upload the repository to GitHub straight away. You’ll need to either purge the large files from the version history; or discard this version history altogether (i.e. remove/rename the .git folder), re-initialize with an updated .gitignore, and git add only the things you want to share.
Step 1b: {here}
Hard-coded paths are common in academic code. Needless to say, they have to go! The main solution in this space is {here}, which basically looks through parent folders recursively until it finds one with an .Rproj file or a .git folder, which indicates that the project root has been located, and then make all paths relative to that.
In other words: read_csv(here("data/input.tsv")) is unambiguous, whereas read_csv("~/Documents/Projects/MPRA/Attempt3/data/input.tsv") only works on one machine, and only if you never move the project folders.
Step 2: {renv} and R
{renv}, the preeminent dependency management for R, was a clear choice. We installed it from Github, initialized it (renv::init()), installed RMarkdown to allow it to parse packages from RMarkdown sources with renv::hydrate(), let it install everything, and then saved it to a lockfile so that we could reproduce it at will (renv::snapshot()).
(I actually made a mistake here — I hadn’t checked what R version Laura had been using, nor have I updated the R that ran on my machine, so we ended up on R 4.0.2 instead of Laura’s R 3.6.1 or the latest R 4.1.1. To my surprise, this presented less of a problem than I thought it would. Still, don’t repeat my mistake! Think of your R version first.)
Why not conda? The main alternative to {renv} was conda with r-forge and bioconda channels. Historically, those have been more painful and often incomplete, plus it doesn’t play nice with RStudio. In hindsight, though, conda was worth attempting, if only to see whether it could make the work with Snakemake easier.
Step 3: Snakemake
For the past several years, I’ve used Makefiles to document my data processing workflows. I’ve read about Snakemake, but I didn’t have time to try it out. Then the time came to help @Laura_Kellman optimize her PhD data processing workflow, so of course I thought now is the time to experiment. (Sorry, Laura.)
To be fair, Snakemake had quite a few features to offer over GNU Make! But I’ve only ever read the docs and haven’t actively deployed it. Rookie mistake.
Aside: the problems with Snakemake
- Snakemake version — the latest version requires using Mamba and mamba-forge, but on Windows, it still defaults to 5.4.0, which is more than an entire version behind. (On OS X, it works correctly, at least, so long as you use Mamba.)
- Snakemake and
{renv}+{RMarkdown}really don’t play nice.script: "X.Rmd"will fail without an explicit error; with the benefit of hindsight, I guess{RMarkdown}/{knitr}was only installed in the{renv}library and so the very first command that Snakemake was trying to execute was failing. It sure feels like it could tell me that, though, instead of failing with a meaningless StopIteration error and nothing at all in the log file! - Snakemake and
{renv}+ bare R don’t play nice, but just in a regular way. By default, Snakemake will call any R script withRscript --vanilla {script}, which intentionally skips loading.Rprofile, which is{renv}’s entire mechanism of action. To counteract that, addsource(".Rprofile")to the top of the scripts. - Snakemake,
{renv}+{RMarkdown}in a separate conda environment? Yeah, couldn’t get that working at all.
So, what worked?
- Snakemake as a sanity check of what goes where. Even if Snakemake didn’t work, writing a
Snakefileclarified where to start and which scripts depend on which. It made it easy to write a bare R script that would run all the notebooks in order, which is basically a low-tech Makefile. - Making scripts Snakemake-ready is a great preparation for deploying them into a computational cluster. We didn’t have time to test it on Sherlock / SCG, but I believe Snakemake would make it easy. (Then again, that’s what I thought at the start about this whole undertaking…)
- Snakemake makes a pretty dependency graph. Look! It’s pretty!
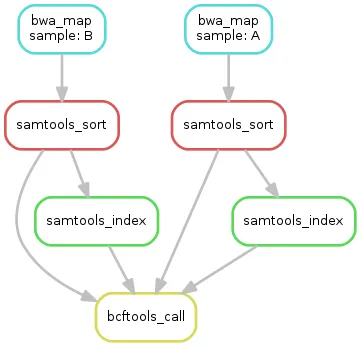
Step 4: Docker (and {renv})
Having a cross-platform replication of the pipeline was already good news, but to make the project run anywhere, we had wanted a Docker container runtime for it.
(If you’ve never heard of Docker, imagine a blank-slate computer that you set up just to run your code. Instead of manually tuning it, you write up every step of the setup in an automated instruction file called the Dockerfile. That way, anyone can set up that same blank-slate computer and anyone can run your code!
…if this isn’t clear, see Boettiger (2014) and Nuest et al. (2020).)
Here were the issues I encountered along the way:
- Docker and
{renv}play games, at least if your plan is to bind-mount the project in at runtime. By default,renv::restore()puts files in yourproject/renv/library/R-{version}/{system/architecture}- which of course won’t work if you’re mountingproject/over it. One way to get around this is to callrenv::isolate(), which appears place your local library in/usr/local/lib/R/site-library- but then you can’t let.Rprofilerun (because it will redirect your.libPaths()to the localrenv/folder), so you have to run your mounted scripts withRscript --vanilla {script}and you can’t explicitly invokesource(".Rprofile")or you’ll break it. (You’ll note that this is, ironically, the exact opposite of what you need to do to work around Snakemake +{renv}interoperability.) - Docker and Snakemake need Singularity to play nice. Singularity is a pain to install on bare Windows; having WSL2 helps.
Steps 5 through N: What we didn’t do, but wanted to
There wasn’t time to do everything. The specific omissions are the following three things in particular:
- Actual code review. We made everything run, and we scrutinized some key variables along the way according to criteria that Laura knew but we didn’t formally codify.
- Automated tests. We didn’t make logical tests with
assertror input/output tests withgreat_expectations. - Cluster deployment. Snakemake promised to make this reasonably easy, but of course by now I’ve learned not to trust Snakemake’s claims. But I’m very excited to try it.
Conclusion
This was unreasonably fun, so much so that I’d consider a job doing nothing but helping bioinformaticians with their pipelines (and briefly looked for it - let me know if you know of one). It was also much, much slower than anticipated, and converted me into a Snakemake skeptic in a way I didn’t anticipate. Looking forward to the next time!
¶Liked this?More writing ↗·Talk to me about it ↗